genomeNLP: Case study of Protein¶
4. Setting up a biological dataset¶
Understanding of the data and experimental design is a necessary first step to analysis. In our case study, we perform a simple two case classification, where the dataset consists of a corpora of biological sequence data belonging to two categories. Protein sequence associated with DNA binding proteins (DBP) and RNA binding proteins (RBP) are available. In the context of biology, DBP interact with DNA molecules, playing roles in gene regulation, DNA replication, repair, and structural organization while RBP interact with various RNA types and function in processes like splicing, RNA stability, translation regulation, and ribosome function. Aberrations in DBP and RBP are implicated in various diseases, including cancer and neurodegenerative disorders. Identifying and classifying these proteins helps in studying disease mechanisms and developing potential therapeutic strategies. Therefore, our aim is to classify sequences into DNA binding protein and RNA binding protein categories.
Our data is available in the form of fasta files. fasta files are a common
format for storing biological sequence data. They typically contain headers that
provide information about the sequence, followed by the sequence itself. They can
also store other nucleic acid data, as well as protein. The fasta format contains
headers with a leading >. Lines without > contain biological sequence data
and can be newline separated. In this example, the full set of characters are
the 20 naturally occurring amino acids Alanine A, Cysteine C, Aspartic Acid D,
Glutamic Acid E, Phenylalanine F, Glycine G, Histidine H, Isoleucine I,
Lysine K, Leucine L, Methionine M, Asparagine N, Proline P, Glutamine
Q, Arginine R, Serine S, Threonine T, Valine V, Tryptophan W and
Tyrosine Y. These are the building blocks of proteins.
#!/bin/bash
# download and preprocess protein data for RNA and DNA binding proteins
# original article: https://doi.org/10.1016/j.jmb.2020.09.008
wget 'http://bliulab.net/iDRBP_MMC/static/dataset/training_dataset.txt'
wget 'http://bliulab.net/iDRBP_MMC/static/dataset/test_dataset_TEST474.txt'
wget 'http://bliulab.net/iDRBP_MMC/static/dataset/test_dataset_PDB255.txt'
csplit --digits=2 --quiet --prefix=outfile training_dataset.txt "/------------------------------------------------------------/+1" "{*}"
sed '$d' outfile02 | sed '$d' > train_dna_binding.fa
sed '$d' outfile04 | sed '$d' > train_rna_binding.fa
rm outfile0*
csplit --digits=2 --quiet --prefix=outfile test_dataset_TEST474.txt "/------------------------------------------------------------/+1" "{*}"
sed '$d' outfile02 | sed '$d' > test_TEST474_dna_binding.fa
sed '$d' outfile04 | sed '$d' > test_TEST474_rna_binding.fa
rm outfile0*
csplit --digits=2 --quiet --prefix=outfile test_dataset_PDB255.txt "/------------------------------------------------------------/+1" "{*}"
sed '$d' outfile02 | sed '$d' > test_PDB255_dna_binding.fa
sed '$d' outfile04 | sed '$d' > test_PDB255_rna_binding.fa
rm outfile0*
# we combine the full dataset and later repartition it with the pipeline
cat train_dna_binding.fa test_TEST474_dna_binding.fa test_PDB255_dna_binding.fa > dna_binding.fa
cat train_rna_binding.fa test_TEST474_rna_binding.fa test_PDB255_rna_binding.fa > rna_binding.fa
gzip dna_binding.fa
gzip rna_binding.fa
The files can be downloaded using the above script. The original publication is accessible here.
HEADER: >Q7YU81
SEQUENCE: MATLIPVNGGHPAASGQSSNVEATYEDMFKEITRKLYGEETGNGLHTLGTPVAQVATSGP
TAVPEGEQRSFTNLQQLDRSAAPSIEYESSAAGASGNNVATTQANVIQQQQQQQQQAESG
NSVVVTASSGATVVPAPSVAAVGGFKSEDHLSTAFGLAALMQNGFAAGQAGLLKAGEQQQ
RWAQDGSGLVAAAAAEPQLVQWTSGGKLQSYAHVNQQQQQQQQPHQSTPKSKKHRQEHAA..
Note
In real world data, other characters are available which refer to multiple possible nucleotides, for example ``W`` indicates either an ``A`` or a ``T``. RNA includes the character ``U``, and proteins include additional letters of the alphabet.
Tokenisation in genomics involves segmenting biological sequences into smaller units, called tokens (or k-mers in biology) for further processing. In the context of proteins, tokens can represent individual amino acids, k-mers or other biologically meaningful segments. Just as in conventional NLP, tokenisation is required to facilitate most downstream operations.
Here, we provide gzipped fasta file(s) as input. While conventional biological tokenisation splits a sequence into arbitrary-length segments, empirical tokenisation derives the resulting tokens directly from the corpus, with vocabulary size as the only user-defined parameter. Data is then split into training, testing and/or validation partitions as desired by the user and automatically reformatted for input into the deep learning pipeline.
Note
We provide the conventional k-merisation method as well as an option for users. In our pipeline specifically, the empirical tokenisation and data object creation is split into two steps, while k-merisation combines both in one operation. This is due to the empirical tokenisation process having to “learn” tokens from the data.
# Empirical tokenisation pathway
$ tokenise_bio -i dna_binding.fa.gz rna_binding.fa.gz -t prot.2000.json -v 2000
# -i INFILE_PATHS path to files with biological seqs split by line
# -t TOKENISER_PATH path to tokeniser.json file to save or load data
# -v VOCAB_SIZE select vocabulary size (DEFAULT: 32000)
This generates a json file with tokens and their respective weights or IDs.
You should see some output like this.
[00:00:00] Pre-processing sequences
[00:00:00] Suffix array seeds
[00:00:14] EM training
5. Format a dataset for input into genomeNLP¶
In this section, we reformat the data to meet the requirements of our pipeline which takes specifically structured inputs. This intermediate data structure serves as the foundation for downstream analyses and facilitates seamless integration with the pipeline. Our pipeline contains a method that performs this automatically, generating a reformatted dataset with the desired structure.
Note
The data format is identical to that used by the HuggingFace ``datasets`` and ``transformers`` libraries.
# Empirical tokenisation pathway
$ create_dataset_bio \
dna_binding.fa.gz \
rna_binding.fa.gz \
prot.2000.json \
-o prot.2000.512 \
--no_reverse_complement \
-c 512
# -o OUTFILE_DIR write dataset to directory as
# [ csv \| json \| parquet \| dir/ ] (DEFAULT:"hf_out/")
# --no_reverse_complement turn off reverse complement (DEFAULT: ON)
# -c CHUNK split seqs into n-length blocks (DEFAULT: None)
# default datasets split: train 90%, test 5% and validation set 5%
The output is a reformatted dataset containing the same information. Properties required for a typical machine learning pipeline are added, including labels, customisable data splits and token identifiers.
DATASET AFTER SPLIT:
DatasetDict ({
train: Dataset ({
features: ['idx', 'feature', 'labels', 'input_ids', 'token_type_ids', 'attention_mask’],
num_rows: 9719 })
test: Dataset ({
features: ['idx', 'feature', 'labels', 'input_ids', 'token_type_ids', 'attention_mask’],
num_rows: 540 })
valid: Dataset ({
features: ['idx', 'feature', 'labels', 'input_ids', 'token_type_ids', 'attention_mask’],
num_rows: 540 })
})
Note
The column ``token_type_ids`` is not actually needed in this specific case study, but it is safely ignored in such cases.
SAMPLE TOKEN MAPPING FOR FIRST 5 TOKENS IN SEQ:
TOKEN ID: 400 | TOKEN: MA
TOKEN ID: 533 | TOKEN: SQS
TOKEN ID: 1742 | TOKEN: EPG
TOKEN ID: 296 | TOKEN: YL
TOKEN ID: 346 | TOKEN: AAA
6. Preparing a hyperparameter sweep¶
In machine learning, achieving optimal model performance often requires finding the right combination of hyperparameters (assuming the input data is viable). Hyperparameters vary depending on the specific algorithm and framework being used, but commonly include learning rate, dropout rate, batch size, number of layers and optimiser choice. These parameters heavily influence the learning process and subsequent performance of the model.
For this reason, hyperparameter sweeps are normally carried out to systematically test combinations of hyperparameters, with the end goal of identifying the configuration that produces the best model performance. Usually, sweeps are carried out on a small partition of the data only to maximise efficiency of compute resources, but it is not uncommon to perform sweeps on entire datasets. Various strategies, such as grid search, random search, or bayesian optimisation, can be employed during a hyperparameter sweep to sample parameter values. Additional strategies such as early stopping can also be used.
To streamline the hyperparameter optimization process, we use the
wandb (Weights & Biases) platform which has a user-friendly interface
and powerful tools for tracking experiments and visualising results.
First, sign up for a wandb account at: https://wandb.ai/site and login by pasting your API key.
$ wandb login
$ wandb: Paste an API key from your profile, and hit enter and hit enter or press ctrl+c to quit :
Now, we use the sweep tool to perform hyperparameter sweep. Search
strategy, parameters and search space are passed in as a json file.
# sweep parameters
{
"method": "random",
"name": "sweep",
"metric": {
"goal": "maximize",
"name": "eval/f1"
},
"parameters": {
"batch_size": {"values": [5, 10, 15]},
"epochs": {"values": [1, 2, 3, 4, 5]},
"learning_rate": {"max": 0.1, "min": 0.0001}
}
}
$ sweep \
prot.2000.512/train.parquet \
parquet \
prot.2000.json \
--test prot.2000.512/test.parquet \
--valid prot.2000.512/valid.parquet \
--hyperparameter_sweep random.json \
--entity_name tyagilab \ # <- edit as needed
--project_name p_sweep \ # <- edit as needed
--group_name prot.2000 \
--output_dir sweep.2000 \
--label_names "labels" \
-n 3
# --test, path to [ csv \| csv.gz \| json \| parquet ] file
# --valid, path to [ csv \| csv.gz \| json \| parquet ] file
# --hyperparameter_sweep, run a hyperparameter sweep with config from file
# --entity_name, wandb team name (if available).
# --project_name, wandb project name (if available)
# --group_name, provide wandb group name (if desired)
# --label_names, provide column with label names (DEFAULT: "")
# -n SWEEP_COUNT, run n hyperparameter sweeps
# -o OUTPUT_DIR, specify path for output (DEFAULT: ./sweep_out)
*****Running training*****
Num examples = 9719
Num epochs= 1
Instantaneous batch size per device = 5
Total train batch size per device = 5
Gradient Accumulation steps= 1
Total optimization steps= 1944
The output is written to the specified directory, in this case
sweep_out and will contain the output of a standard pytorch
saved model, including some wandb specific output.
The sweeps gets synced to the wandb dashboard along with various
interactive custom charts and tables which we provide as part of our
pipeline. A small subset of plots are provided for reference.
Interactive versions of these and more plots are available on wandb.



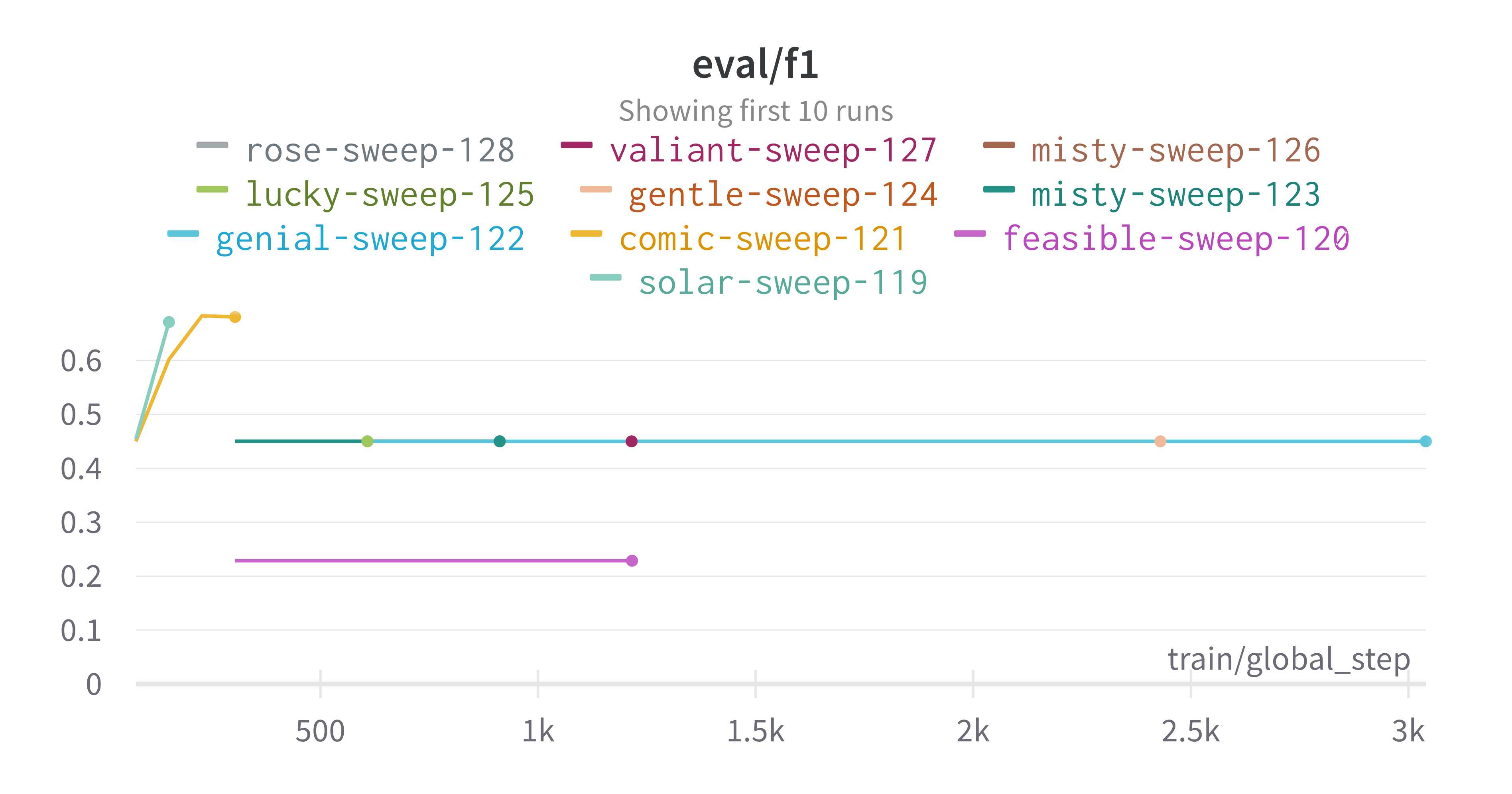

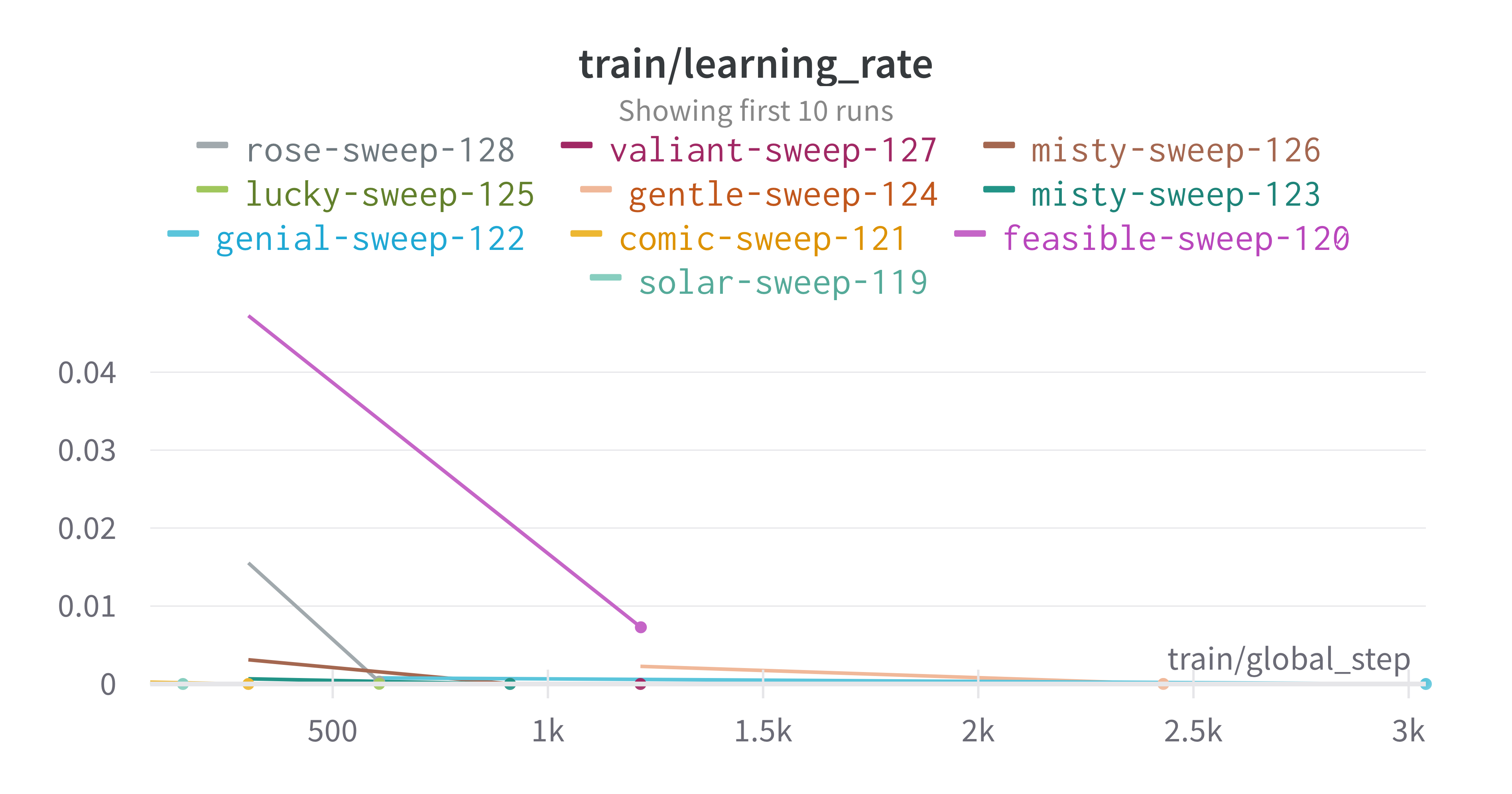
Here is an example of a full wandb generated report:
You may inspect your own generated reports after they complete.
7. Selecting optimal hyperparameters for training¶
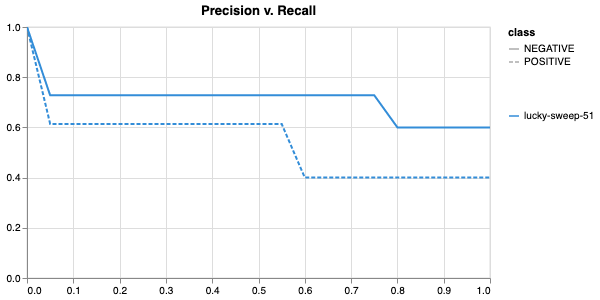
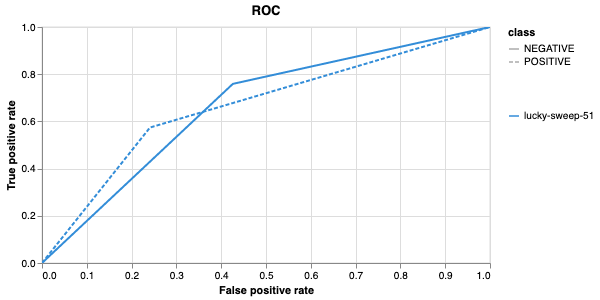
Having completed a sweep, we next identified the best set of parameters for model training. We do this by examining training metrics. These serve as quantitative measures of a model’s performance during training. These metrics provide insights into the model’s accuracy and generalisation capabilities. We explore commonly used training metrics, including accuracy, loss, precision, recall, and f1 score to inform us of a model’s performance
A key event we want to avoid is overfitting. Overfitting occurs when a
learning model performs exceptionally well on the training data but
fails to generalise to unseen data, making it unfit for use outside of the
specific scope of the experiment. This can be detected by observing performance
metrics, if the accuracy decreases and later increases an overfit
event has occurred. In real world applications, this can
lead to adverse events that directly impact us, considering that such
models are used in applications such as drug prediction or self-driving cars.
Here, we use the f1 score calculated on the testing set as the main
metric of interest. We showed that we obtain a best f1 score of 0.677488189237731.
Best run kind-sweep-18 with eval/f1=0.677488189237731
BEST MODEL AND CONFIG FILES SAVED TO: protein_sweep/model_files
HYPERPARAMETER SWEEP END
You may inspect your own generated reports after they complete.
8. With the selected hyperparameters, train the full dataset¶
In a conventional workflow, the sweep is performed on a small
subset of training data. The resulting parameters are then
recorded and used in the actual training step on the full dataset.
Here, we perform the sweep on the entire dataset, and hence
remove the need for further training. If you perform this on your
own data and want to use a small subset, you can do so and then
pass the recorded hyperparameters with the same input data to
the train function of the pipeline. We include an example of
this below for completeness, but you can skip this for our
specific case study. Note that the input is almost identical to
sweep.
$ train \
prot.2000.512/train.parquet \
"parquet" \
prot.2000.json \
--test prot.2000.512/test.parquet \
--valid prot.2000.512/valid.parquet \
--entity_name tyagilab \
--project_name prot \
--group_name train.2000 \
--config_from_run tyagilab/prot/2niwyeqs \
--output_dir train.out \
--label_names "labels" \
--overwrite_output_dir
# -t TEST, path to [ csv \| csv.gz \| json \| parquet ] file
# -v VALID, path to [ csv \| csv.gz \| json \| parquet ] file
# -w HYPERPARAMETER_SWEEP, run a hyperparameter sweep with config from file
# -e ENTITY_NAME, wandb team name (if available).
# -p PROJECT_NAME, wandb project name (if available)
# -l LABEL_NAMES, provide column with label names (DEFAULT: "").
# -n SWEEP_COUNT, run n hyperparameter sweeps
The contents of hyperparams.json, the file with the best hyperparameters identified by the sweep.
{
"output_dir": "./sweep_out/random",
"overwrite_output_dir": false,
"do_train": false,
"do_eval": true,
"do_predict": false,
"evaluation_strategy": "epoch",
"prediction_loss_only": false,
"per_device_train_batch_size": 32,
"per_device_eval_batch_size": 32,
"per_gpu_train_batch_size": null,
"per_gpu_eval_batch_size": null,
"gradient_accumulation_steps": 1,
"eval_accumulation_steps": null,
"eval_delay": 0,
"learning_rate": 0.00000017248305228664,
"weight_decay": 0.5,
"adam_beta1": 0.9,
"adam_beta2": 0.999,
"adam_epsilon": 1e-08,
"max_grad_norm": 1.0,
"num_train_epochs": 2,
"max_steps": -1,
"lr_scheduler_type": "linear",
"warmup_ratio": 0.0,
"warmup_steps": 0,
"log_level": "passive",
"log_level_replica": "passive",
"log_on_each_node": true,
"logging_dir": "./sweep_out/random/runs/out",
"logging_strategy": "epoch",
"logging_first_step": false,
"logging_steps": 500,
"logging_nan_inf_filter": true,
"save_strategy": "epoch",
"save_steps": 500,
"save_total_limit": null,
"save_on_each_node": false,
"no_cuda": false,
"use_mps_device": false,
"seed": 42,
"data_seed": null,
"jit_mode_eval": false,
"use_ipex": false,
"bf16": false,
"fp16": false,
"fp16_opt_level": "O1",
"half_precision_backend": "auto",
"bf16_full_eval": false,
"fp16_full_eval": false,
"tf32": null,
"local_rank": -1,
"xpu_backend": null,
"tpu_num_cores": null,
"tpu_metrics_debug": false,
"debug": [],
"dataloader_drop_last": false,
"eval_steps": null,
"dataloader_num_workers": 0,
"past_index": -1,
"run_name": "./sweep_out/random",
"disable_tqdm": false,
"remove_unused_columns": false,
"label_names": null,
"load_best_model_at_end": true,
"metric_for_best_model": "loss",
"greater_is_better": false,
"ignore_data_skip": false,
"sharded_ddp": [],
"fsdp": [],
"fsdp_min_num_params": 0,
"fsdp_transformer_layer_cls_to_wrap": null,
"deepspeed": null,
"label_smoothing_factor": 0.0,
"optim": "adamw_hf",
"adafactor": false,
"group_by_length": false,
"length_column_name": "length",
"report_to": [
"wandb"
],
"ddp_find_unused_parameters": null,
"ddp_bucket_cap_mb": null,
"dataloader_pin_memory": true,
"skip_memory_metrics": true,
"use_legacy_prediction_loop": false,
"push_to_hub": false,
"resume_from_checkpoint": null,
"hub_model_id": null,
"hub_strategy": "every_save",
"hub_token": "<HUB_TOKEN>",
"hub_private_repo": false,
"gradient_checkpointing": false,
"include_inputs_for_metrics": false,
"fp16_backend": "auto",
"push_to_hub_model_id": null,
"push_to_hub_organization": null,
"push_to_hub_token": "<PUSH_TO_HUB_TOKEN>",
"mp_parameters": "",
"auto_find_batch_size": false,
"full_determinism": false,
"torchdynamo": null,
"ray_scope": "last",
"ddp_timeout": 1800
}
The output is written to the specified directory, in this case
train_out and will contain the output of a standard pytorch
saved model, including some wandb specific output.
The trained model gets synced to the wandb dashboard along with
various interactive custom charts and tables which we provide as part
of our pipeline. A small subset of plots are provided for reference.
Interactive versions of these and more plots are available on wandb.



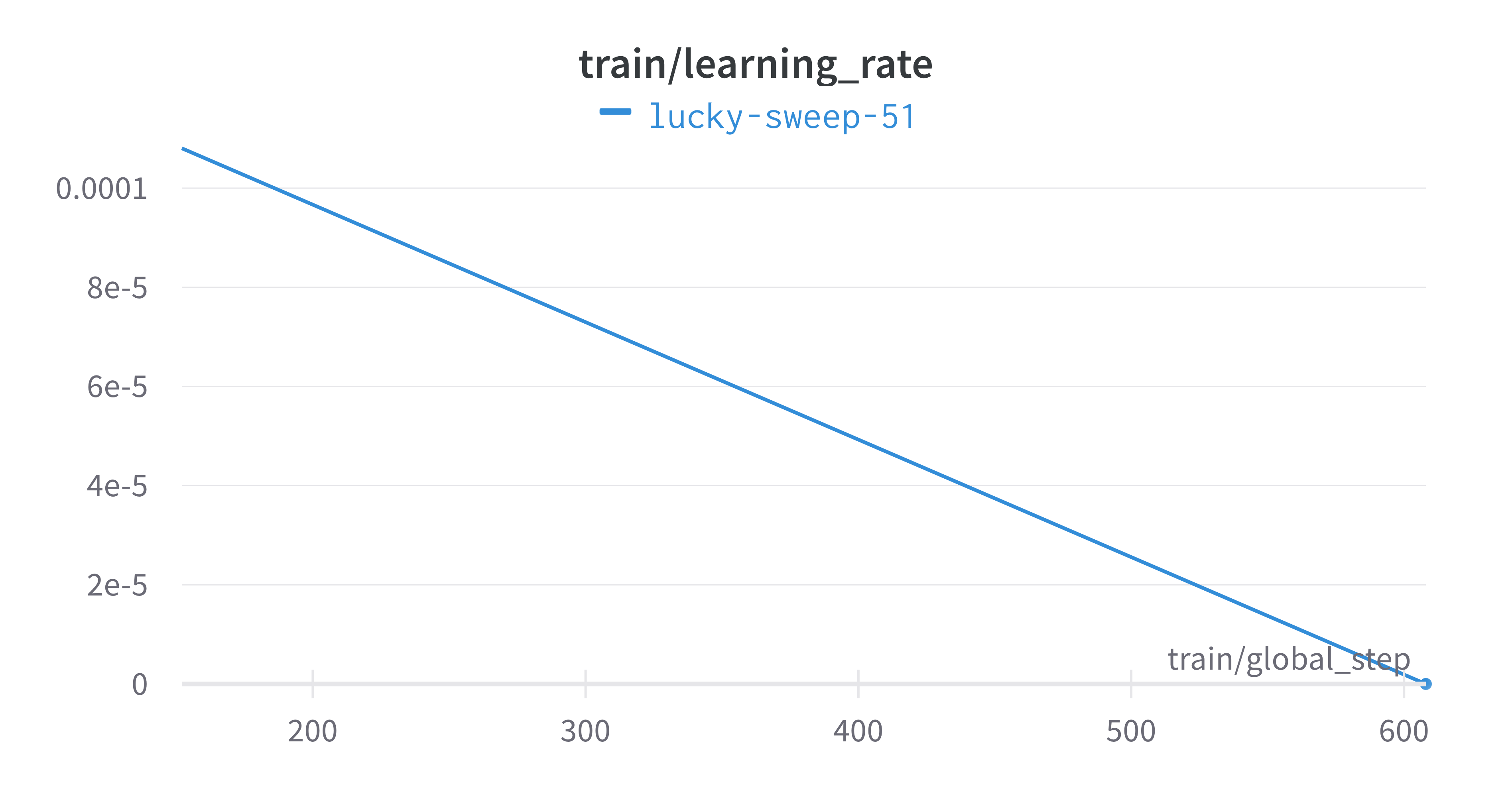
Here is an example of a full wandb generated report:
You may inspect your own generated reports after they complete.
9. Perform cross-validation¶
Having identified the best set of parameters and trained the model, we next want to conduct a comprehensive review of data stability, and we do this by evaluating model performance across different data slices. This assessment is known as cross-validation. We make use of k-fold cross-validation in which data is divided into k subsets and the model is trained and tested on these individual subsets.
$ cross_validate \
data.csv/train.parquet parquet \
-t data.csv/test.parquet \
-v data.csv/valid.parquet \
-e tyagilab \
-p testm3 \
--config_from_run p9do3gzl \ # id of best performing run
--output_dir cv \
-m sweep_out \
-l labels \
-k 3
# --config_from_run WANDB_RUN_ID, *best run id*
# –-output_dir OUTPUT_DIR
# -l label_names
# -k KFOLDS, run n number of kfolds
*****Running training*****
Num examples = 8504
Num epochs= 4
Instantaneous batch size per device = 64
Total train batch size (w, parallel, distributed & accumulation)= 64
Gradient Accumulation steps= 1
Total optimization steps= 532
Automatic Weights & Biases logging enabled
The cross-validation runs are uploaded to the wandb dashboard along
with various interactive custom charts and tables which we provide as
part of our pipeline. These are conceptually identical to those generated
by sweep or train. A small subset of plots are provided for reference.
Interactive versions of these and more plots are available on wandb.

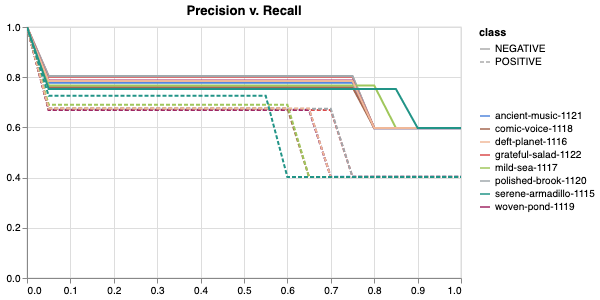
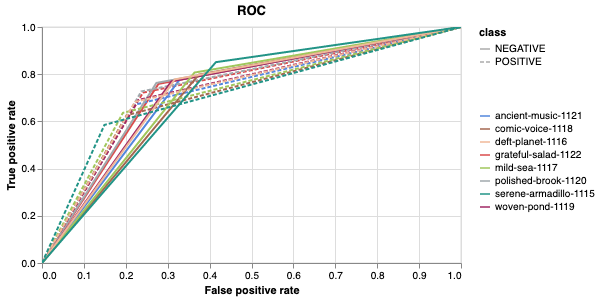
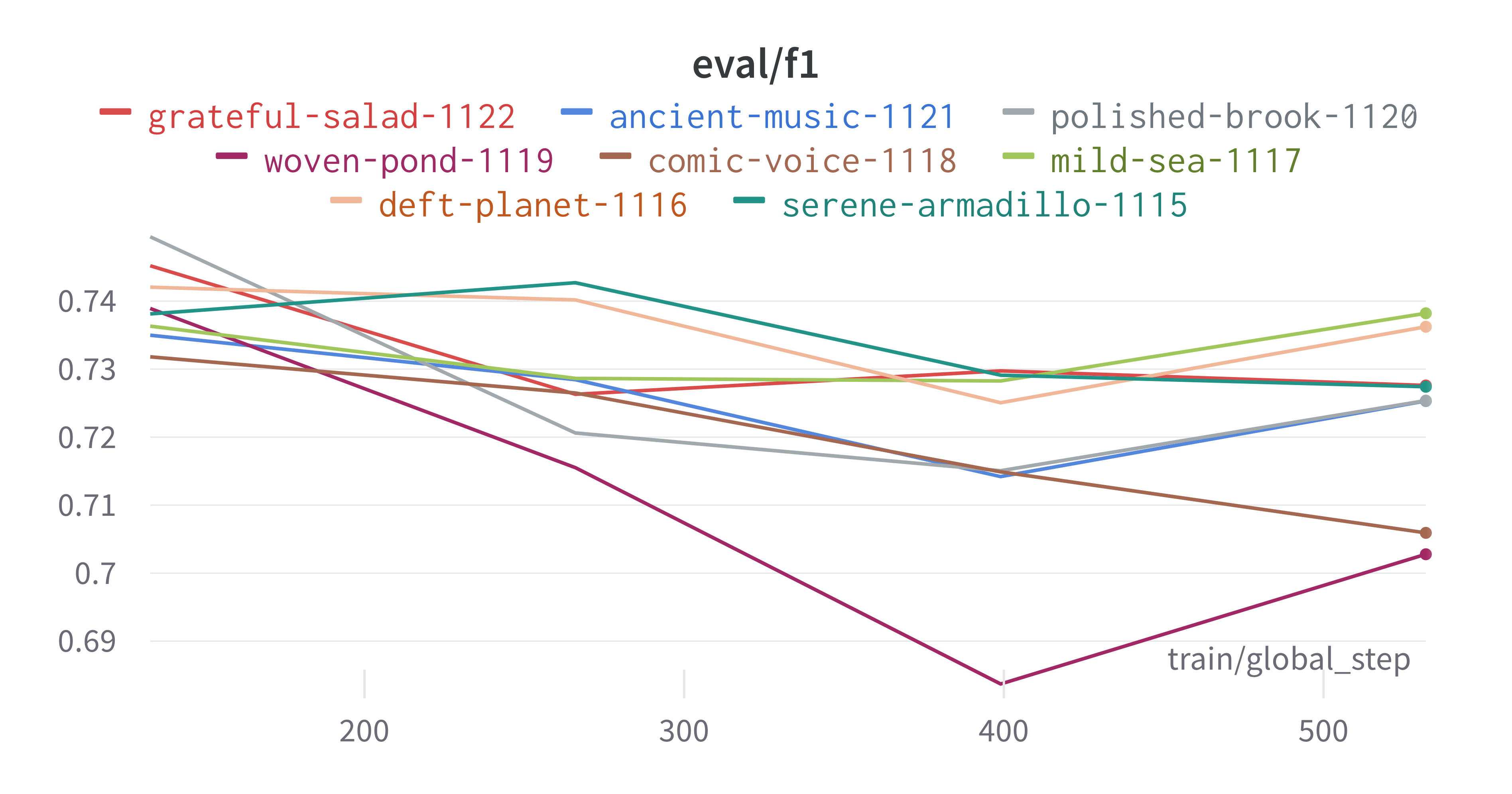
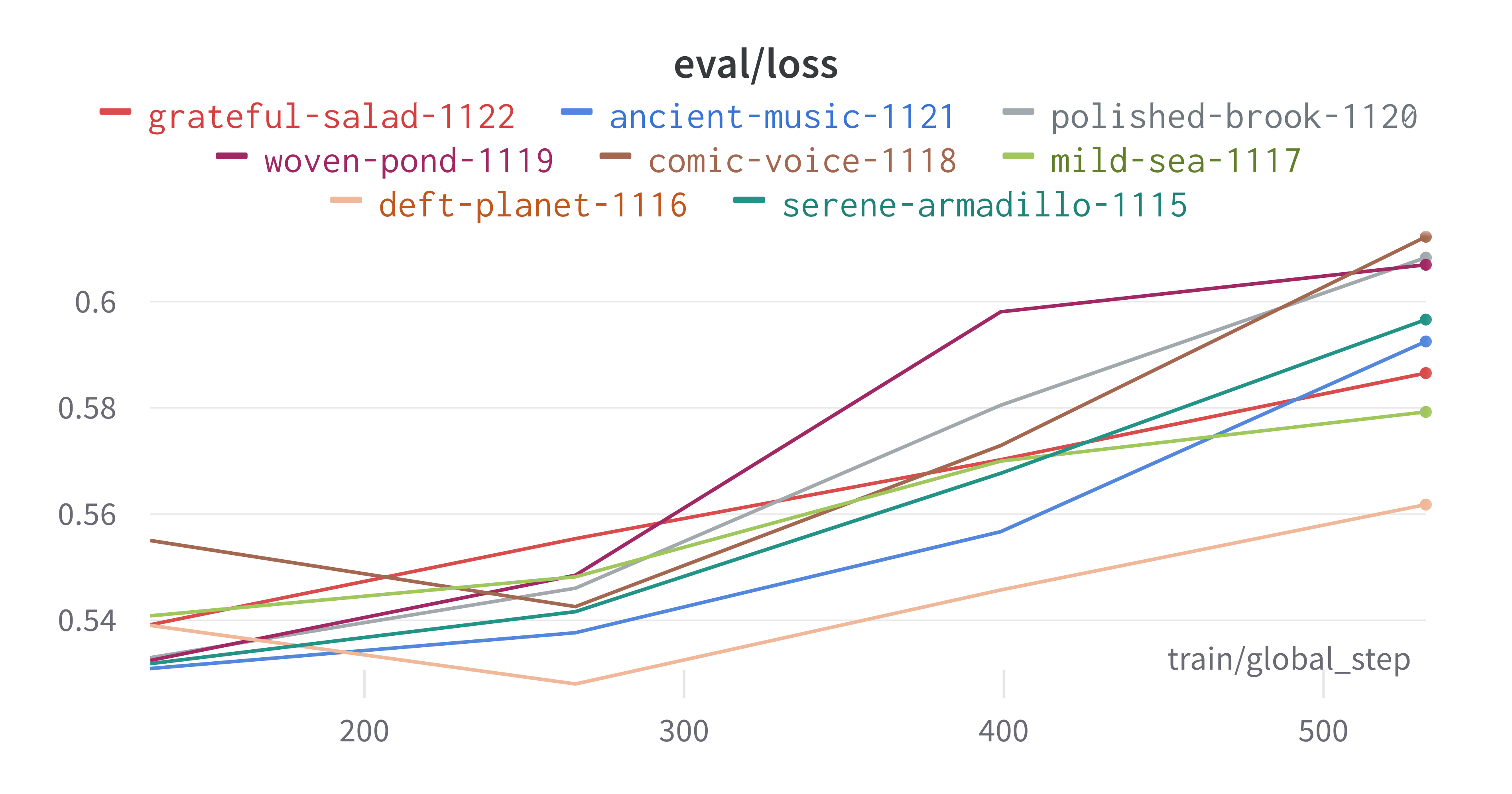

Here is an example of a full wandb generated report:
You may inspect your own generated reports after they complete.
10. Compare different models¶
The aim of this step is to compare performance of different deep learning models efficiently while avoiding computationally expensive re-training and data download in conventional model comparison. In the case of patient data, they are often inaccessible for privacy reasons, and in other cases they are not uploaded by the authors of the experiment.
For the purposes of this simple case study, we compare multiple sweeps of the same dataset as a demonstration. In a real life application, existing biological models can be compared against the user-generated one.
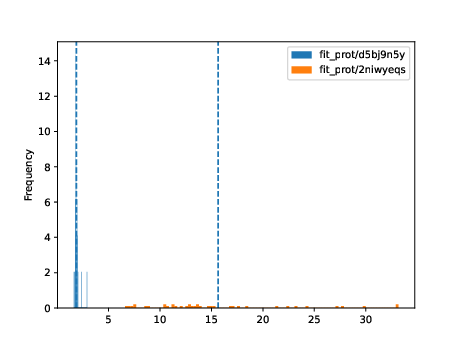
$ fit_powerlaw tyagilab/prot/d5bj9n5y tyagilab/prot/2niwyeqs -o fit_prot
# -m MODEL_PATH, path to trained model directory
# -o OUTPUT_DIR, path to output metrics directory
This tool outputs a variety of plots in the specified directory.
$ ls fit_prot
> alpha_hist.pdf alpha_plot.pdf model_files/
Very broadly, the overlaid bar plots allow the user to compare the performance of different models on the same scale. A narrow band around 2-5 with few outliers is in general cases an indicator of good model performance. This is a general guideline and will differ depending on context! For a detailed explanation of these plots, please refer to the original publication.

11. Obtain model interpretability scores¶
Model interpretability is often used for debugging purposes, by allowing the user to “see” (to an extent) what a model is focusing on. In this case, the tokens which contribute to a certain classification are highlighted. The green colour indicates a classification towards the target category, while the red colour indicates a classification away from the target category. Colour intensity indicates the classification score.
In some scenarios, we can exploit this property by identifying regulatory regions or motifs in DNA sequences, or discovering amino acid residues in protein structure critical to its function, leading to a deeper understanding of the underlying biological system.
$ gzip -cd dna.binding.fa.gz | head -n22 > dna_subset.fasta
$ interpret tyagilab/prot/d5bj9n5y dna_subset.fasta -o prot_interpret
# -o OUTPUT_DIR, specify path for output
Citation¶
Cite our manuscript here:
@article{chen2023genomicbert,
title={genomicBERT and data-free deep-learning model evaluation},
author={Chen, Tyrone and Tyagi, Navya and Chauhan, Sarthak and Peleg, Anton Y and Tyagi, Sonika},
journal={bioRxiv},
month={jun},
pages={2023--05},
year={2023},
publisher={Cold Spring Harbor Laboratory},
doi={10.1101/2023.05.31.542682},
url={https://doi.org/10.1101/2023.05.31.542682}
}
Cite our software here:
@software{tyrone_chen_2023_8135591,
author = {Tyrone Chen and
Navya Tyagi and
Sarthak Chauhan and
Anton Y. Peleg and
Sonika Tyagi},
title = {{genomicBERT and data-free deep-learning model
evaluation}},
month = jul,
year = 2023,
publisher = {Zenodo},
version = {latest},
doi = {10.5281/zenodo.8135590},
url = {https://doi.org/10.5281/zenodo.8135590}
}